Solución a los problemas sobre purificación de proteínas.
Esta sección contiene 7 problemas.
----------regresar al índice general------------
1.- a) Para Separar las proteínas por medio de electroforesis es necesario usar un soporte que permita que las moléculas separadas no se mezclen nuevamente y puedan identificarse mediante alguna técnina de revelado, como puede ser la tinción con azul ce Coomasie. Para ello se emplean soportes como la Agarosa y la Acrilamida/bisacrilamida. En el caso de las porteínas, el soporte mas conveniente es este último. La malla del gel permite que las moléculas al avanzar, empujadas en el campo eléctrico, gracias a su carga eléctrica, sean "frenadas" en función de su tamaño y forma. De modo que la electroforesis en gel permite separar moléculas mediante un acombinación de su carga y de su masa.
Ahora, debido a que queremos separar las subunidades, debemos debilitar las interacciones covalentes y no covalentes que las mantienen unidas, tales como los punetes de disulfuro, las interacciones hidrofóbicas, los puentes de hidrógenos, las interaciones iónicas y las llamadas fuerzas de Van der Walls. Para ello basta con agragar un detergente, que reemplaza las interacciones hidrofóbicas proteína-proteína, por interacciones detergente-proteína, al hacer esto, expone al solvente gran parte del interior de la proteína y esto permite que se formen puentes de hidrógeno con el agua y que se solvaten los grupos cargados. Para romper los puentes de disulfuro que pudiesen existir, es necesario reducir estos enlaces para formar dos sulfhidrilos. Los compuestos tales como el 2-mercaptoetanol (2ME), el 2,3-dimercaptopropanol (BAL), el dithioeritritol (DTE), el dithitreitol (DTT), y el Tris (2-carboxietil) fosfito (TCEP), son reductores que pueden emplearse bajo diferentes condiciones, siendo el primero el más débil (se emplea en caliente) y el último el más fuerte. En estas condiciones, la carga negativa del SDS hace casi uniforme la relación carga/masa de las proteínas, de modo que la separación depende casi enteramente del tamaño y la forma.
En resumen, necesitamos Acrfialmida/biscarilamida, Azul de Coomasie, 2-mercaptoetanol y SDS.
b) Para partir una cadena polipeptídica en fragmentos pueden emplearse métodos químicos o métodos enzimáticos, que pueden romper enlaces peptídicos en los que participan aminoácidos específicos. Entre los primeros está el Bromuo de cianógeno (CNBr) y entre los segundos se encuentras las endoproteinasas como la quimotrispsina.
c) Si desaemos digerir una cadena hasta aminoácidos para luego determinar la composición de la misma, podemos emplear HCL 6N @ 90 oC, lo que a lo largo de varias horas (24-72h) resultará en una ruptura completa de los enlaces peptídicos que forman el péptido. Los aminoácidos pueden analizarse separandolos mediante cromatografía de fase reversa (HPLC), cromatografía de intercambio iónico ó cromatografía de capa fina. Los picos de aminoácidos pueden detectarse mecdiante su reacción con el reactivo de ninhidrina. Para evitar que los grupos SH se oxiden a disulfuros, y dificulten la digestión, se emplea ácido perfórmico para oxidarlos a ácido cisteico o iodoacetamina, para formar el derivado S-alquílico correspondiente.
d) Finalmente, si lo que se desea es determinar la secuencia de aminoácidos de la proteína, debe emplearse un método que permita digerir uno a uno los amináciods. Esto puede hacerse mediante la degradación de EDMAN, que emplea el fenilisotiocianato y determina las feniltiohidantoinas de los aminoácidos en el extremo aminoterminal, como él péptido digerido es ahora un aminoácido más corto, al repetirse el proceso se determina el segundo aminoácido y así sucesivamente.
----------regresar al índice de la sección -------------------regresar al índice general------------
2.- En la electroforesis desnaturalizante, el logaritmo del peso molecular observa una relación aproximadamente lineal (negativa) con la mobilidad relativa. Haciendouna gráfica semilogarítmica del peso de los marcadores vs. su mobilidad relativa, se puede interpolar el valor del peso molecular de la lisozima, que según se ve en la figra siguiente es de 26 KDa.
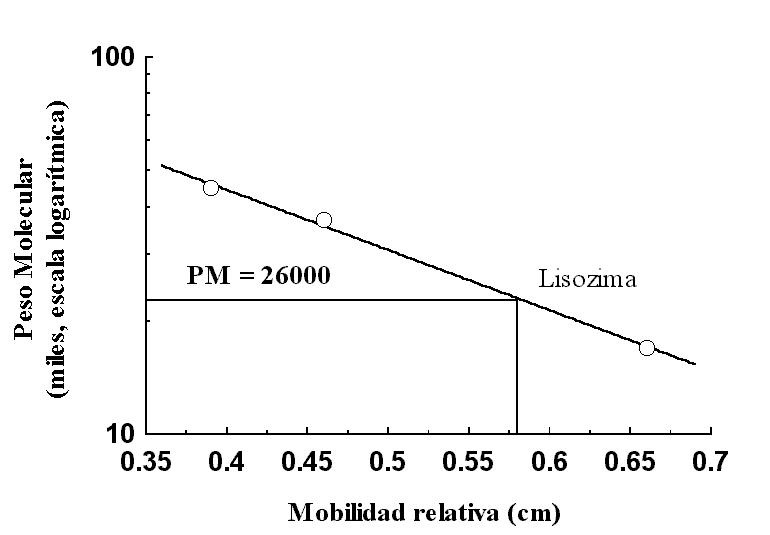
----------regresar al índice de la sección -------------------regresar al índice general------------
3.- a) En este problema se nos pide completar la tabla de purificación añadiendo las columnas de actividad específica, las veces de purificación y el rendimiento obtenido en cada paso. Aquí, hay que ver que significan las dos columnas que nos dan y que información se nos pide.
Primero, la columna de "actividad total" expresa cuanta de la enzima que se desea purificar está presente en cada paso; la segunda columna expresa cuantas proteínas de entre el total de ellas, la que nos interesa incluída, están siendo recobradas con cada paso de purificación.
Es evidente que deseamos obtener lo más posible de la acetilcolinesterasa, al tiempo que recobremos tan pocas de las otras proteínas como sea posible. Así, la actividad específica es el cociente entre la actividad total y la cantidad total de proteína presente en esa preparación. Este valor será mayor si se recupera más acetilcolinesterasa y menos de otras proteínas. Para la acetilcolinesterasa 100% pura, este valor alcanzará un número constante, ya que al no haber contaminantes ya no será posible eliminar proteína sin que desaparezca la actividad correspondiente a la enzima perdida. El valor se obtiene símplemente dividiendo el valor de la celda en la columna 1 por el valor de la columna 2 correspondiente a la misma fila (vease la tabla más abajo).
Luego, para calcular las veces de purificación, bastará considerar que entre más pura esté la proteína, mayor su actividad específica, de manera que si dividimos la actividad específica recuperada en cada paso por la que encontramos en la preparación original, sabremos cuantas veces se ha enriquecido nuestra preparación en colinesterasa, con respecto a otras porteínas. Es decir dividimos el valor de cada celda en la columna 3 de la tabla inferior, por el valor de la primera celda en esa misma columna, evidentemente el valor de la primera fila será igual a la unidad, porque ahí la enzima conserva la pureza correspondiente al extracto original, puesto que no ha sido tratada.
Finalmente, la última columna corresponde al rendimiento, es decir cuanto del compenente que se intenta purificar se está recobrando en cada paso, aquí basta referirnos a la actividad total recuperada de la columna 1, ya que este es el valor que cuantifica componente deseado. Así, expreando el valor de cada una de estas celdas como porcentaje del valor presente en el extracto crudo (es decir la fila 1 de esta columna), se genera la columna 5.
|
Paso de purificación |
Actividad total (µmol/min) |
Proteína total (µg) |
Actividad específica µmol/(min mg) |
Veces de purificación |
Rendimiento (%) |
|
Homogenizado de tejido fresco. |
556.7 |
33340 |
1.67 |
1 |
100 |
|
Precipitación con (NH4)2SO4 |
556.7 |
1070 |
52.03 |
31.16 |
100 |
|
DEAE-celulosa |
289.5 |
124 |
233.47 |
139.82 |
52 |
|
concentración y díalisis |
278.4 |
115 |
242.09 |
144.98 |
50.01 |
|
sephadex-G200 |
233.8 |
56 |
417.5 |
250.04 |
42 |
|
Cellex-P (intercambio catiónico) |
139.2 |
55.4 |
251.26 |
150.48 |
25 |
|
DEAE-celulosa |
89.1 |
11.3 |
788.5 |
472.22 |
16.01 |
|
DEAE-sephadex |
66.8 |
11.1 |
601.8 |
360.41 |
12 |
|
cromatografía de afinidad |
46.8 |
4.8 |
975 |
583.91 |
8.41 |
b y c) Al analizar la tabla se observa que hay varios pasos en los que se obtiene un rendimiento razonable con respecto al paso anterior y además hay una ganacia en la veces de purificación. En particular, La precipitación con sulfato de amonio mejora la pureza 31 veces y nos dá un rendimiento del 100%, la DEAE celullosa permite incrmentar la pureza, aunque se pierde bastante actividad. La concentración y diálisis es un paso que no añade nada, pero tampoco se pierde la proteína y que es necesario para poder aplicar la muestra a la siguiente columna. El paso de filtación en gel por sephadex G200 mejora la pureza y mantiene un buen rendimiento. El paso de intercambio catiónico siguiente, es malo, porque se pierde bastante actividad y se recupera con menos pureza de la que se tenía. La siguiente cromatografía repite el paso de DEAE-cellulosa, que a pesar de todo representa una mejora con respeto a la proteína obtenida del paso de filtración, sin embargo, otro paso de más intercambio aniónico (DEAE-suphadex, mismo grupo químico, diferente soporte inerte) no mejora más la pureza, sino que incluso hay cierta pperdida de pureza y de actividad. Finalmente, el paso de cromatografía de afinidad si es bueno, ya que se recupera una proteína 900 veces más pura que la original y el rendimento pasa tan sólo de 12 a 8.4 %, es decir casi 60% de lo aplicado a este último paso. En resumen, sería conveniente eliminar los pasos de Cellex-P y DEAE-sephadex (letra magenta, fondo amarillo), con lo cual, en teoría, sería posible obtener una proteína de pureza semejante, con menos trabajo y con una mejora en el rendimiento final.
----------regresar al índice de la sección -------------------regresar al índice general------------
4.- La respuesta a este problema se puede dar a partir de la siguiente consideración: En un campo electroforético las moléculas nativas de proteína se moveran en hacia el electrodo de cargo opuesta a la propia, de modo que se elegimos un pH con intermedio entre el punto isoeléctrico de ambas proteínas, cada una se moverá hacia un polo diferente, aquella con un pI inferior al pH poseerá carga negativa y se moverá hacia el ánodo (polo positivo) y aquella con el pI superior al pH tendrá carga negativa y se moverá al cátodo (polo negativo). De este modo, al moverse en direcciones opuestas, la separación será óptima. En el tercer caso, como se trata de 3 proteínas, podemos elegir un pH igual al pI de la que tenga un valor intermedio, entonces, una se moverá hacia el cátodo, otra hacia el ánodo y la de pI intermedio no se moverá.
a) Seroalbúmina pI 4.5 y Hemoglobina pI 6.8, pH recomendado 5.65
b) Mioglobina pI 7 y Quimotripsinógeno pI 9.6, pH recomendado 8.3
c) Ovoalbúmina pI 4.5 , Seroalbúmina pI 4.9 y Ureasa pI 5, pH recomendable 4.9. En relidad esta es un pregunta capciosa, ya que debido a la cercanía entre los puntos isoeléctricos de la seroalbúmina y la ureasa, esta serían muy difíciles de separar, pero el valor óptimo para el pH seguiría siendo 4.9.
----------regresar al índice de la sección -------------------regresar al índice general------------
5.- En la cromatografía de exclusión molecular, las proteínas son separadas en función de su peso molecular. Las moléculas de mayor tamaño son excluídas de la resina y son eluídas con poco volumen, pero las de menor peso molecular son retenidas y se requiere de mayor volumen de eluyente para lavarlas. Por tanto el orde de elución, comenzando por la que eluyen primero sería: Miosina (525), catalasa (222), seroalbúmina (69), quimotripsinógeno (23), Mioglobina (17), citocromo c (13). El númeor entre paréntesis indica el peso molecular el miles de daltons.
----------regresar al índice de la sección -------------------regresar al índice general------------
6.- Este problema se puede resolver de manera semejante al problema 2, ya que el logaritmo del peso molecular varía linealmente (pero en forma negativa) con el volumen de elución. El resultado se muestra en la figura siguiente, en la que se vé que la Lisozima posee un peso molecular aproximado de 24 KDa en su forma nativa.

----------regresar al índice de la sección -------------------regresar al índice general------------
7.- El problema 5 nos dice que la lisozima posee un peso molecular en forma nativa de 24 KDa y en su forma desnaturalizada el peso molecular fue de 26 KDa. La diferencia puede atribuirse a la impresición de los métodos y también al hecho de que una proteína desnaturalizada, al estar más expuesta al solvente, tiene una forma más "abultada" y sufre mayor fricción con el gel y el solvente en su movimiento electroforético. De modo que podemos concluir que la lisozima es una proteína monomérica con un peso de 24-26 Kda, o sea que no posee estructura cuaternaria.
----------regresar al índice de la sección -------------------regresar al índice general------------
Solución a los problemas sobre el comportamiento cinético de las enzimas.
Esta sección contiene 11 problemas.
1.- En este problema se nos pide analizar como progresa la inactivación de una enzima en el tiempo. Aquí, la ciclohexanodiona (CHD) es un reactivo que modifica químicamente con la enzima y el producto de esta reacción es una proteína que pierde su actividad enzimática original. Por tanto, en este experimento, la enzima es un reactante y su actividad enzimática es el recurso metodológico que sirve para evaluar la concentración remanente de proteína no modificada por la CHD, en cada uno de los tiempos muestreados.
Se puede determinar si la reacción química es de primer orden, o se encuentra en condiciones tales que progresa como si lo fuese (aunque estríctamente no lo sea); ello gracias a que la reacciones con cinética de primer orden (o de pseudoprimer orden) muestran una relación lineal entre el logaritmo natural de la concentración de reactante remanente y el tiempo transcurrido. De modo que, si obtenemos los logarítmos de los valores de concentración de la enzima remanente (no importa que se trate de un porcentaje) resultan las siguientes gráficas.
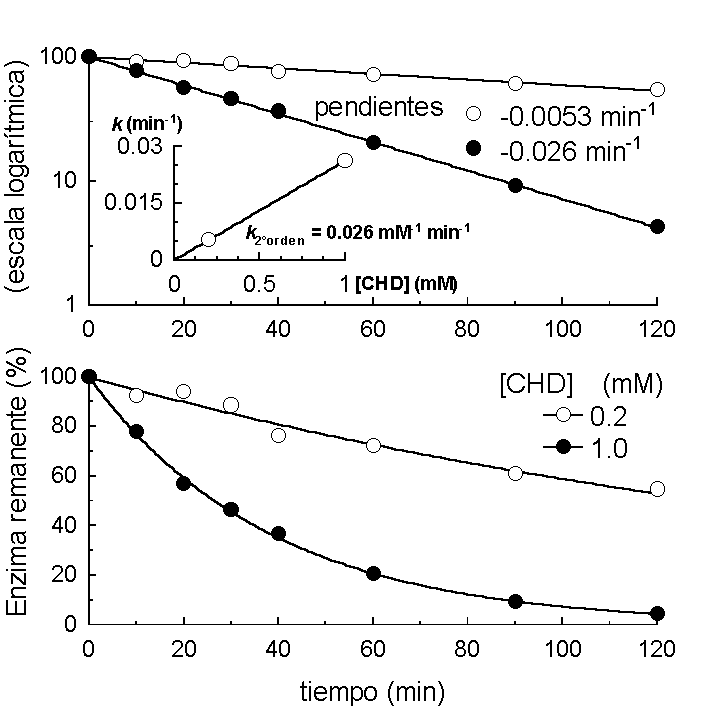
La figura superior muestra la gráfica logarítmica, en la que se puede observar que ambos conjuntos de puntos describen razonablemente una recta, no así en la gráfica directa de [E] vs. tiempo (panel inferior). Este resultado indica que la inactivación observada sigue una cinética de primer orden con respecto a la concentración de enzima en la condiciones empleadas. Las pendientes de estas líneas son numéricamente iguales a las constantes de primer orden, o de pseudoprimerorden, pero con signo negativo, de modo que basta cambiar el signo para determinar el valor de las constantes buscadas.
Note que ambas rectas presentan pendientes muy distintas, esto significa que la concentración de CHD si tiene un efecto sobre la velocidad de la reacción annque, en las condiciones experimentales empleadas, este efecto no se manifiesta en los 120 minutos que dura el experimento; posíblemente porque este reactivo se encuentra en gran exceso. Sin embargo, una expresión correcta para la velocidad de la reacción debería incluir ambos reactantes y la ley de velocidad indicaría que el orden de reacción es superior a 1 (uno para la enzima y no sabemos exactamente cuanto para la CHD).
Finalmente, el recuadro en la figura superior muestra que las constantes aparentes calculadas varían linealmente con la cantidad de CHD empleada en el experimento y la línea cruza el punto x,y (0,0). Esto nos indica que el orden de reacción para la CHD es también de 1 y la pendiente de esta gráfica nos indica el valor de la constante de 2o orden. y la expresión final para la velocidad sería:
v= k2 [CHD][E], en donde k2= 0.026 mM-1 min-1 = 0.43 M-1 s-1.
En condiciones en las que [CHD] es mucho mayor que [E], la enzima se agota sin que la [CHD] disminuya apreciablemente. Esto es muy frecuente en la inactivación de enzimas, ya que la concentración de proteína apenas alcanza los cientos de mg/mL, lo que para una molécula de 10000 gr/mol de PM (si se trata de na proteína pequeña) o más, representa concentraciones inferiores a 10-5 M. Por su parte el agente químico suele agragarse en concentraciones superiores a 10-4M, es decir 10 veces por encima como mínimo. Concencuentemente, el cambio en concentración de reactante ráramente rebaza el 10% a lo largo de la incubación.
----------regresar al índice de la sección -------------------regresar al índice general------------
2.- Para resolver este problema es necesario observar que la absorbencia registrada representa la desaparición del NADH, que es el sustrato de la reacción. En este caso la estequimetría de la reacción implica que por cada evento de reacción, una molécula de NADH desaparece y, por tanto, lo que necesitamos hacer para calcular la velocidad es determinar el Cambio de absorbencia del NADH por unidad de tiempo, para luego convertirlo a cambio de concentración de NADH mediante la ley de Lambert-Beer:
|
A = e c l |
|
Dc = DA/(e l) |
|
ó |
de donde se sigue que |
ó |
|
DA = e Dc l |
|
Dc/Dt = (DA/Dt)/(e l) |
Si graficamos la absorbencia contra el tiempo transcurrido, la pendiente de la curva descrita por la gráfica será (y2-y1)/(x2-x1) = DA/Dt. La que en este caso será numéricamente negativa (por tratarse de la desaparición de un producto). Sin embargo, podremos notar en la figura inmediata inferior, que la relación no describe una recta perfecta, por lo que es necesario considerar tan sólo el periodo inicial, en el que las concentraciones de sustratos y productos no han cambiado apreciablemente y, por tanto, los punto se aproximan razonablemente a una recta.
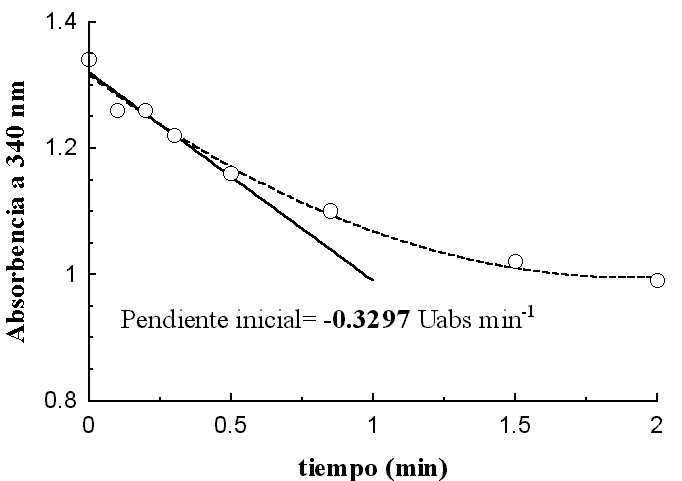
Así, con una pendiente inicial de -0.3297 Uabs/min y una longitud para el paso de luz de la celda del espectrofotómetro de 1 cm (l) se obtiene una velocidad de desaparición de NADH de 5.3´10-5 M min-1, lo que en 3 mL del volumen de cubeta representa (5.3´10-5 mol L-1 min-1) ´ (3´10-3 L) = (1.59´10-7 mol min-1).
Las unidades internacionales (UI) de actividad enzimática son mmoles de producto formado o reactante consumido por minuto, de modo que si 1 mol equivale a 106 mmol, la velocidad será (v = 1.59´10-1 mmol min-1) ó 0.159 UI.
Finalmente, la actividad específica es l actividad enzimática dividida por la concentración de proteína, es decir a.e. = 0.159 UI/35 ng, pero debido a que la concentración de proteína suele expresarse en mg: a.e. = 0.159 UI / 35´10-6 mg = 4543 UI/mg de proteína.
----------regresar al índice de la sección -------------------regresar al índice general------------
3.- En este problema se nos pide determinar gráficamente la relación entre la velocidad de la reacción catalizada, vs. la cantidad de solución de proteína añadida al ensayo y luego se nos pide calcular el cociente de la velocidad (ó actividad de la enzima) y la cantidad de solución deproteína añadida al ensayo. Los resultados de tal representación se muentran en la siguiente figura:
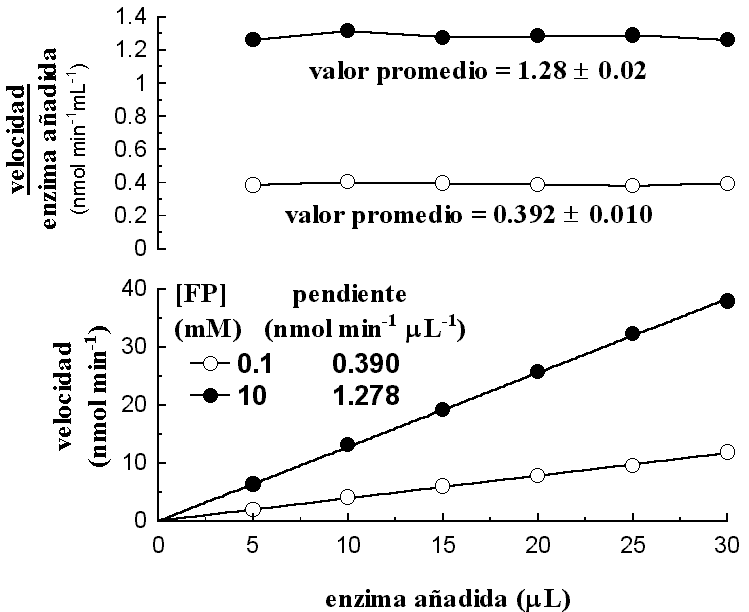
En el panel inferior, apreciamos que la velocidad aumenta linealmente con la concentración de proteína, esto es lo que cabría esperar, debido a que, si tomamos como referencia la ecuación de Michaelis-Menten, notaremos que la velocidad es proporcional a la Velocidad máxima y a su vez la Vmax es proporcional a la concentración total de enzima añadida, i.e.:
![]()
Dado que en cada una de las dos series de determinaciones la concentración de sustrato se mantuvo constante, el factor kCAT S / (KM + S) se mantiene constante y, por tanto, podemos decir que mientras se empleé la misma concentración de sustrato en todas las medidas, v = constante ´ ET. Consecuentemente, cuando se grafica el cociente v/[E] el resultado es una constante (vease panel superior de la figura anterior), mientras la concentración de sustrato no sea modificada.
Lo anterior significa que la velocidad de una reacción enzimática es proporcional a la concentración de enzima, lo que constituye la base teórica de las mediciones de actividad enzimática como una estimación de la cantidad de esta enzima presente en un fluído biológico. Este principio tiene, por ejemplo, mucha importancia en la práctica clínica, ya que nos permite determinar alteraciones en los niveles de diversas enzimas, que pueden relacionarse a diversos estados patológicos. También es importante en aplicaciones en la industria de alimentos, ya que permite evaluar la cantidad de ciertas enzimas presentes en los alimentos, cuya actividad puede ser indeseable, o bien, puede ser añadida ex professo para modificar las propiedades de los preparados alimenticios. Todo ello, mediante un ensayo enzimático, generalmente sencillo, que tan sólo requiere como condición que la concentración de sustrato empleada, así como otras condiciones (pH, temperatura, etc.), sean estandarizadas adecuadamente.
----------regresar al índice de la sección -------------------regresar al índice general------------
4.- En este problema se nos dá el valor de la velocidad como función de la cantidad de sustrato añadido a tres diferentes niveles de concentración de enzima. El valor de Vmax y Km puede calcularse a partir de estos datos de diversas maneras. La manera directa, aunque no muy precisa, consiste en graficar los datos y estimar el valor de Vmax a partir del valor al que tiende la velocidad cuando la concentración de sustrato es muy elevada. Como en realidad este valor es el límite superir de la curva, pero esta se aproxima indefinidamente a él sin llegar a tocarlo, estimar este techo resulta un poco impresiso. Con el valor obtenido, es posible calcular el valor de la Km, ya que este representa la concentración de sustrato a la que 50% de los sitios activos de la enzima están ocupados con sustrato y 50% están vacíos. En estas condiciones, la velocidad observada deberá ser igual a la Vmax/2. (véase panel A de la figura que sigue a este párrafo). Como se describió en el problema anterior, aumentar la concentración de enzima resulta en un incremento en la Vmax, ya que v = VMAX´ET. Por tanto, las tres curva son semejantes en su forma, pero a mayor cantidad de enzima tenemos curvas más elevadas. Como es de esperar, cuando dividimos la velocidad por la concentración de proteína empleada, la tres curvas se superponen (ver panel B de la figura), dado que aquí la velocidad ya sólo dependerá de la concentración de sustrato. Esta es una manera común de representar las curvas de saturación por su sustrato de las reacciónes enzimáticas, ya que no su forma es la misma sin importar cuanta enzima se empleó en el experimento.

Los páneles de la derecha en la figura anterior (C,D) son representaciones de Linewaver-Burk, en esta representación se grafica 1/velocidad vs. 1/[sustrato], por lo que también se le conoce como la gráfica de dobles recíprocos. Los valores de 1/v frente a la 1/S describen una línea recta, cuya pendiente es numéricamente igual a KM/VMAX y que intesecta al eje "Y" en un valor numérico igual a 1/VMAX y si la recta se prolonga a través del cuarto cuadrante intersecta al eje "X" en un valor numérico igual a -1/KM. En la figura, el panel derecho inferior (C) muestra que la KM es independiente de la concentración de enzima añadida. Esto puede deducirse del hecho de que la concentración de enzima es casi siempre mucho menor que la de él sustrato (vease problema 1), por lo que la cantidad de sustrato requerida para ocupar el 50% de los sitios activos sólo depende de la afinidad relativa entre la enzima y el sustrato, propiedad que no cambia con la concentración total de enzima. Esto mismo puede deducirse de la ecuación de Michaelis-Menten, ya que en ella, la concentración de ET y la de sustrato no son interdependientes. El panel superior dereho (D) reitera que al dividir la velocidad por la concentración de enzima empleada las tres líneas se superponen.
Si ahora graficamos velocidad máxima obtenida contra la cantidad de enzima añadida en mmol, asumiendo que cada molécula de enzima posee un sólo sitio activo, la pendiente de la gráfica será:
![]()
Este valor nos dice que una molécula de enzima es capáz de convertir 33300 moléculas de sustrato a poducto por segundo y es equivalente a la actividad específica de una enzima pura (VMAX/[Proteína]), excepto por la unidades empleadas. Dado que para determinar este último número no necesitamos una estimación exacta del peso molecular de la enzima, no del número de sitios activos por molécula de proteína, este valor es el más frecuentemente reportado en diversas tablas de parámentros cinéticos y propiedades de enzimas. Sin embargo, cuando la preparación no es pura el denominador de este conciente será mayor, debido a la contribución de los contaminantes a la cantidad total de proteína, en consecuencia, la actividad específica disminuirá. De este modo, la actividad específica es un buen indicador del grado de pureza de una preparación enzimática.
----------regresar al índice de la sección -------------------regresar al índice general------------
5.- En este problema se nos pide determinar la actividad como fracción de la VMAX para la enzima invertasa, la manera más simple de proceder es reordenar la ecuación de Michalis y Menten como sigue:
![]()
Lo que nos indica que se alcanza el 33% de la VMAX, como VMAX = kCAT ´ ET , se tiene vel = 175 min-1 ´ 2.5 ´ 10-6 mmol ´ 0.33 = una actividad de 0.00146 mmol min-1. Ahora, según lo abtenido arriba, para obtener un 95 % de la VMAX bastará con que S/(KM + S) = 0.95, de donde al despejar S tendremos S= KM ´ 0.95/(1-0.95) = 19 KM. Lo que significa que habrá que añadir 19 x 0.5 mM = 9.5 mM de sacarosa para que la actividad alcance este valor.
Este problema sugiere que la cantidad de sustrato es uno de los factores importantes a considerar cuando se desea emplear una enzima para aplicaciones inductriales. En otras palabras, si el producto a tratar, que en este caso podría ser un almibar hecho a base de sacarosa, posee un cancentración de sustrato muy baja, o sea que nuestro jarabe está muy diluído, deberemos añadr mucha más enzima, puesto que ésta estaría trabajando muy por debajo de su capacidad máxima. Por otro lado, una alternativa para ahorrar enzima, sería tratar el jarabe concentrado y luego añadir el agua y los otros componentes de su almibar, para tener el producto final.
----------regresar al índice de la sección -------------------regresar al índice general------------
6.- En este problema se pide calcular KM y VMAX para una enzima que posee dos sustratos. En este caso, podemos variar la cancentración de oxoglutarato y mantener la concentración de aspartato constante y viceversa. El problema consiste en saber que concentración de aspartato debo emplear cuando varío el oxoglutatato y que concentración de oxoglutaroto es recomendable mantener, al variar el osoglutarato. Aquí, existen dos valores para la KM, dado que este valor representa la cocentración de sustrato que ocupa 50% de los sitios activos de la enzima en el estado estacionario, sin embargo, se pueden ocupar estos sitios activos con ambos sustratos y, por tanto, habrá una KM(oxoglutarato ) y otra KM(aspartato).
La figura sigiente muestra la gráfica de velocidad contra concentración de sustrato (panel izquierdo, para los 3 experimentos) y la de 1/v vs 1/s (panel derecho).
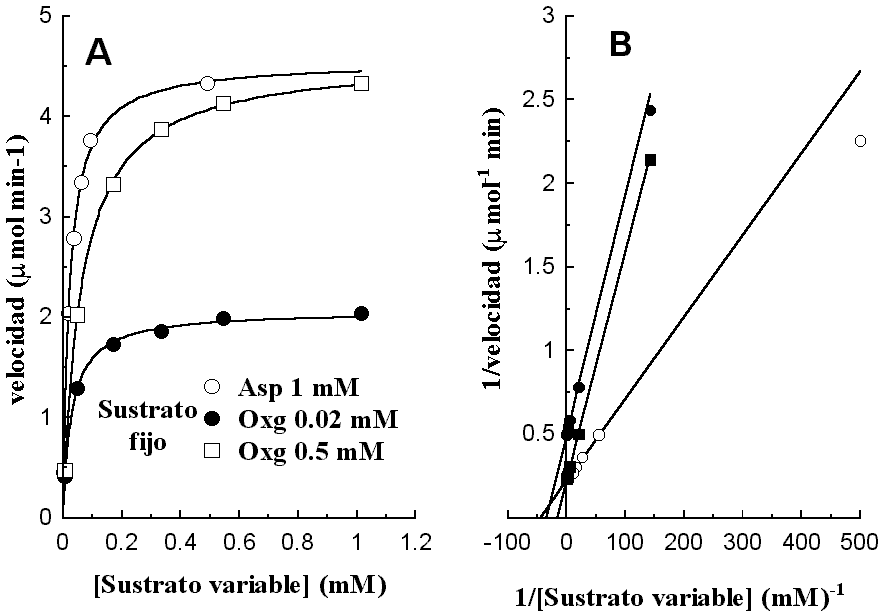
Puede observarse que las curvas y las rectas correspondientes a los experimentos 1 y 3 extrapolan a la misma VMAX, este es el primer diagnóstico. La VMAX es la velocidad de catálisis cuado la enzima se encuentra saturada con sus sustrato, pero dado que aquí hay dos sustratos, habrá que saturar con ambos. Esto significa que cuando variamos la concentración de aspartato, la concentración de oxoglutarato debe estar saturante desde el primero hasta el último tubo medido y viceversa. Si esto no se asegura, entonces el resultado será como el mostrado en el experimento 2, en el que la VMAX determinada es considerablemente inferior al valor real, decimos que se trata de un valor aparente. Ahora -¿cómo sabemos que la concentración de un sustrato es o no saturante?- bueno, como se vió en el problema anterior, nosotros nos aproximamos al 95% de la VMAX cuando la concentración de sustrato es 19 veces KM. En este caso, habrá que elegir una concentración de oxoglutarato igual a 19*KM(oxoglutarato ) cuando se varía el aspartato y viceversa.
El problema es que de entrada, no sabemos cual es el valor de la KM para ninguno de los sustratos y, por tanto, no es posible determinar a priori que concentración será saturante. Para resolver este dilema se realiza un primer experimento y se determina el valor de KM para un sustrato (el que se varió). En nuestro caso, se trata del experimento con oxoglutarato variable. Con este resultado, se calcula el valor de KM(oxoglutarato ) aparente y se realiza entonces otro variando la concentración de aspartato con una concentración de osoglutarato de al menos 20*KM(oxoglutarato ). De este nuevo experimento se determina ahora la KM(aspartato), con lo que podemos saber si la concentración de aspartato empleada anteriormente fue realmente saturante. Si la respuesta es afirmativa, ambos valores de VMAX deben ser muy cercanos. De lo contrario, es necesario repetir el primer experimento con una concentración de sustrato mayor a la empleada inicialmente.
En nuestro caso, la
KM(oxoglutarato) determinada en el primer
experimento es 0.022 mM y la VMAX 4.54 mmol
min-1. En el segundo experimento, realizado con 0.02 mM
de oxoglutarato, la concentración no fue realmente saturante
y no es sorprendente ver que el valor de Vmax es inferior al
anterior (2.06 mmol min-1)
y la KM(aspartato) obtenida no es necesariamente
el valor real tampoco (0.03 mM). Sin embargo, en el tercer
experimento, la concentración de oxoglutarato fue de 0.5 mM,
es decir, 22 veces la KM(oxoglutarato) del primer
experimento (que por cierto, aún no sabemos si es cercana al
valor real), por consiguiente la VMAX es ahora
mayor (4.58 mmol min-1,
de hecho más cercana a la obtenida en el primer experimento)
y el valor de KM(aspartato) es ahora 0.062 mM.
Es
decir, ahora podemos determinar que la concentración de
aspartato empleada en el primer experimento fue 16 veces
KM(aspartato), cercano a la saturación,
pero se deseamos resultados aún más limpios, habría
que repetir este experimento con una concentración aún
mayor (digamos 2 mM).
----------regresar al índice de la sección -------------------regresar al índice general------------
7.- En este problema se nos pide calcular la energía de activación de Arhenius, para ello, el procedimiento más sencillo es el obtener una grafíca de Log(k) vs. 1/T (absoluta) y la pendiente de esta grafica será igual a -Ea/R (siendo R la constante universal de los gases). En este caso no tenemos el valor de k, sino tan sólo el valor de la velocidad de reacción. Sin embargo, la pendiente de una gráfica de Arhenius es:
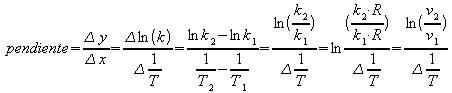
Ello gracias a que la velocidad es proporcional a la concentración de reactante (v = k R) y asumiendo que ésta no se modificó durante el experimento. Lo que significa que para efectos del cálculo de la pendiente y de la energía de Activación, es equivalente graficar la k o la velocidad directamente, siempre que las mediciones se hallan realizado con un cantidad constante de reactante.
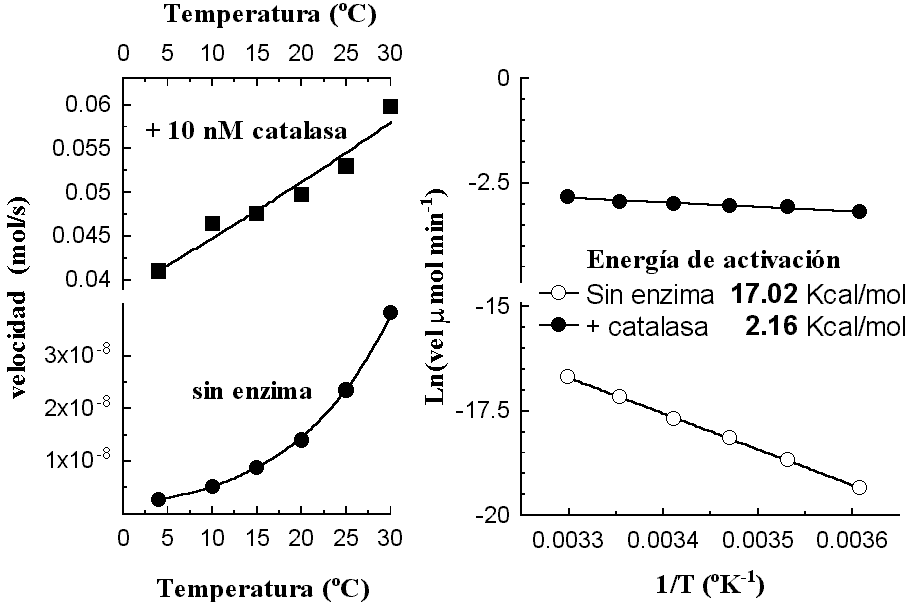
La fugura anterior muestra la gráfica directa de velocidad vs. Temperatura, la que muestra que la velocidad de la reacción crece exponencialmente con la temperatura. También es claro, que en el caso de la reacción catalizada, no se observa una caída en el rango de temperatura empleado, lo que nos dice que no se presenta desnaturalización térmica de la proteína y, por ello, es posible emplear todos los puntos para construir el gra´fico de Arhenius, mismo que se muestra a la derecha de la figura anterior. Note que para realizar esta igura las temperaturas fueron primero convertidas a temperaturas absolutas en oK.
De la pendiente de la gráfica y sabiendo que R=1.987 cal mol-1 oK-1 se obtienen los valores para la energía de Activación de la reacción catalizada por la enzima y en ausencia de catalizador, ya que EA = pendiente ´ -1 ´ R. Aquí se puede ver que la energía de activación de la reacción catalizada por la enzima es cerca de 8 veces menor que la de la reacción en ausencia de enzima. Lo que confirma que las enzimas actúan reduciendo el tamaño de esta barrera enegética.
----------regresar al índice de la sección -------------------regresar al índice general------------
8.- Este es un problema en el que lo que tenemos que hacer es muy semejante a lo que hicimos en el problema anterior. Aquí sin embargo hay que determinar que datos debemos usar. Dado que la gráfica de Arhenius implica la relación entre Log(k) vs 1/T absoluta, y k es una constante de velocidad, debemos identificar aquí la constante de velocidad relacionada con el proceso de catálisis, esta el la kCAT. La KM como puede verse, también puede ser modificada por la temperatura, debido a que su valor es una relación de varias constantes de velocidad (vease la definición de KM en la deducción de la ecuación de Michaelis y Menten), por tanto, al cambiar la temperatura las constantes de velocidad cambián y esto afecta el valor de KM. Esto es lógico, ya que la temperatura también puede afectar la unión del sustrato a la proteína y este proceso se refleja en el valor de KM.
Ahora, en adición a lo anterior, se puede identificar en la siguiente figura que a las temperaturas más altas se observa un claro efecto de desnaturación de la proteína y por tanto una caida en la catálisis. Dado que la energía de activación que deseamos medir es la del proceso catalítico, debemos considerar sólamente los puntos que reflejan el efecto de la temperatura sobre la reacción catalizada y no acusan aún efectos sobre la reacción química paralela de desnaturalización de la proteína:
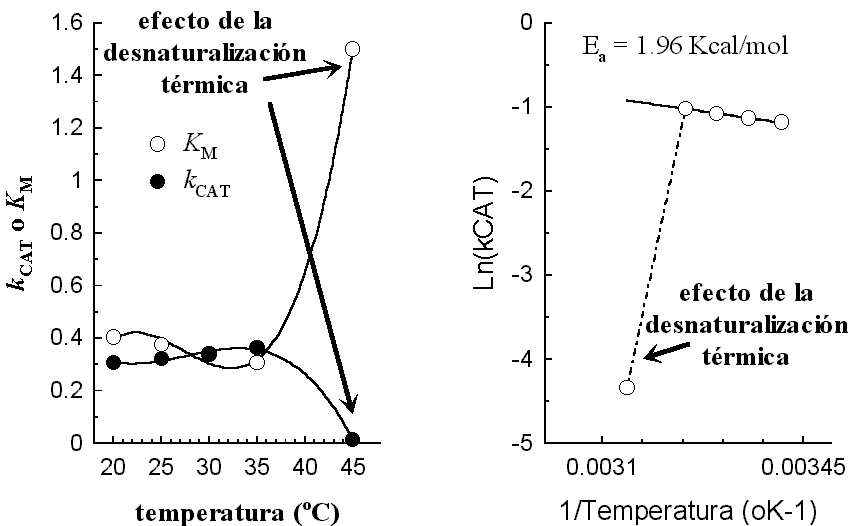
En la gráfica de la derecha, se puede ver la recta que se obtiene en el gráfico Ln(k) vs 1/T, del que se deduce una energía de activación de 1.96 Kcal/mol. Nótese como cláramente el punto que corresponde a la mayor temperatura no cae sobre la recta. Esto debido a que este punto presenta una mezcla de los efectos de la temperatura sobre la velocidad de catálisis y sobre la velocidad de desnaturalización de la proteína.
----------regresar al índice de la sección -------------------regresar al índice general------------
9.- En este problema se nos pide determinar el valor del pKa del grupo involucrado en la respuesta de la enzima frente al pH. Así, haciendo una gráfica de la actividad frente al pH, se obtiene la curva siguiente (línea contínua):

Esta curva es equivalente a una curva de titulación como las que se ven cuando se añade base a un ácido debil en solución y se siguen los cambio de pH, sólo que aquí, en lugar de equivalentes de base añadida, lo que estamos modificando el el pH y observando los equivalentes de enzima activa presentes. Dado que el intervalo de pH empleado no es muy extremo, es de esperarse que la caída en la actividad hacia el pH de 6, se reflejo de que la mayoría de la enzima no está en su forma iónica catalíticamente competente, por lo que no manifiesta su actividad, pero es improbable que esté irreversiblemente desnaturalizada. En cualquier caso, para estar completamente seguros de este hecho, habría que hacer un experimento distinto, en el que la enzima se incubaría a los diferentes pH durante un cierto intervalo de tiempo y luego se regresaría al pH de 8.5-9 (en donde tiene su máxima actividad de acuerdo con la curva anterior) y se mediría cuanta de la enzima puede recuperarse después del tratmiento. Este experimento nos daría la misma actividada a todos los pH's en los que la enzima es estable y reportaría bajas de la actividad en las regiones en las que el pH desnaturaliza a una fracción de la proteína en forma irreversible.
a) A partir de la curva dibujada, se puede ver que el punto de inflexión de la curva es paroximadamente de 6.7, lo que se da a un valor de activida de aproximadamente la mitad del máximo observado a un pH entre 8.5 y 9.
b) Lo anterior nos indica que podría tratarse de algún aminoácido como la histidina. Menos probables, pero no se pueden descartar, serían los carboxilatos de Asp y Glu o el grupo SH de una cisteína libre (es decir que no esté formando puente de disulfuro. Aunque los pKa de los grupos R de estos últimos 3 aminoácidos en su forma libre se encuentran relativamente alejados de 7 (4.7 para los carboxilatos y 8 para el tiol), en el interior de las proteínas y especialmente en el sitio activo de las enzimas, se pueden observar desviaciones impertantes de los pKa, causadas por las características de polaridad del sitio activo, que son casi seimpre muy diferentes a las del medio acuoso puro.
c) Fnalmente, la línea punteada de la figura anteríor indica de manera aproximada lo que se espera observar a valores de pH mucho más alcalinos. En esas condiciones, la desprotonación masiva de muchos aminoácidos, es decir casi todas las histidinas, los tioles y la gran mayoría de lisinas, así como muchas argininas, perderán su carga positiva provocando la desestabilización de numerosos puentes de sal en la proteína, con lo que la estructura se verá severamente afectada. Consicuentemente, se espera a que un pH superior a 10 u 11, la actividad comenzará a desiminuir abruptamente y para un pH de 12 o 13 la enzima presentará una actividad nula. Esto es casi siempre cierto, pero cabe aclarar que algunas proteínas de bacterias alcalofílicas y que se encuentran en medios extremos pueden soportar pH cercanos a 14 sin perdida de la actividad.
----------regresar al índice de la sección -------------------regresar al índice general------------
10.- parte a. En este problema, podemos graficar los valores de velocidad frente a la concentración de sustrato para las series obtenidas sin y con las distintas concentraciones del inhibidor. Esto se muestra en la figura que sigue, en el panel inferior izquierdo. Aunque es posible estimar los valores de Km y Vmax (en ausencia del inhibidor) y los valores de las constantes aparentes en presencia del inhbibidor en esta figura, el estimado será difícil de hacer, debido a que la Vmax es una valor límite que debe extrapolarse y el valor de Km debe estimarse una vez que se conoce la Vmax, lo que significa que un mal estimado de Vmax dará un mal estimado de Km. Una manera sencilla de resolver este problema es realizar la gráfica que se presenta en el lado izquierdo de la figura, la representación de 1/v vs. 1/s propuesta por Linewaver & Burk permite, uniéndo los puntos con una recta, determinar los valores de 1/Vmax a partir de la intercepción con el eje ordenado y -1/Km a partir de la intercepción de la recta extrapolada al eje de las abscisas. Aunque existen otras formas de graficar estos datos que pueden ser más convenientes desde el punto de vista del estimado numérico obtenido, esta es la más extendida en la literatura.
a) A partir de la representación mostrada en el panel derecho de la figura, puede también determinarse el tipo de inhibidor que se tiene, que en este caso es acompetitivo (también llamado incompetitivo), dado que las rectas que unen cada una de las series de puntos son paralelas. Note que aquí de poco sirve emplear un análisis de regresión lineal, dado que los errores experimentales provocan que las rectas obtenidas por regresión no presenten la misma pendiente. En estos casos, una vez que se ha decidido que los puntos realmente representan rectas paralelas, lo mejor es tomar una regla y trazar todas las recta paralelas buscando que el conjunto de líneas realmente describan, tan cercánamente como sea posible, el conjunto de puntos experimentales.
b) Aquí, el único valor de Vmax para la enzima es el determinado en ausencia del inhibidor (O). Es decir 1/Vmax = 0.965 (mmol-1 min mg prot) ó Vmax = 1.04 (mmol min-1 mg prot-1). Los valores obtenidos con las otras rectas representan tan sólo parámetros cinéticos aparentes cuyo valor refleja las alteraciones impuestas sobre la actividad enzimática por la presencia del inhibidor. Km se determina por consiguiente del valor de la intercepción con las absisas de esta misma recta i.e. -1/Km = -2.4 mM-1 ó Km = -1/-2.4 mM = 0.417 mM de p-nitrofenilfosfato. Esto significa que cuando la concentración de p-nitrofenilfosfato en el medio de reacción (al pH y temperatura empleados en este experimentos) alcanzan 0.417 mM, el 50% de los sitios activos de la enzima se encontrarán saturados y por tanto la actividad derá igual a la mitad de la máxima que puede observarse en dichas condiciones.

c) Finalmente, dado que los inhibidores acompetitivos afectan al valor de la intercepción con la ordenada de la gráfica de (1/v,1/s), los valores de este parámetro pueden emplearse para calcular gráficamente el valor de Ki sin recurrir al álgebra. Para ello, basta graficar estos valores frente a la concentración de inhibidor empleada en cada serie de determinaciones, lo que se muestra en el panel izquierdo superior. Aquí, se unen los puntos con una recta y se extrapolan al eje de las absisas, el punto de crte es numéricamente igual a -Ki = -20.47 mM ó Ki = 20.47 mM de bromolevamisol. Note que el primer punto de la gráfica representa el valor de la intercepción cuando la concentración de inhibidor el cero, es decir, este punto es numéricamente igual a 1/Vmax.
parte b. a) La solución de este problema es muy semejante a la descrita para el problema anterior, de manera que no abundaremos en los detalles. Aquí, el patrón del gráfico de Lineweaver-Burk es claramente no paralelo, sin embargo, habrá que dterminar si las rectas se cortan en un punto sobre el eje de las ordenadas, o sobre un punto en el segundo o tercer cuadrante. La distinción entre ambos casos debe hacerse nuevamente al observar todo el conjunto de puntos y el empleo de la regresión lineal aporta poco para una elección adecuada del conjunto de rectas y del punto de corte entre ellas. En el caso presente es claro que el mejor patrón es un abanico de rectas que se cortan en el eje de las ordenadas, por lo que puede decirse con facilida que el inhibidor es competitivo. Note que a pesar de tener la misma Vmax, las curva de la gráfica directa no dan la apariencia de tener la misma asíntatota. Esto se debe a que en cada una de las series de experimentos, al estar una concentración de inhibidor distinta presente, se alcanza un grado de saturación distinta de la enzima y por ello es muy difícil decidir si todas las rectas, dada una concentracion de sustrato suficientemente alta, alacanzarán el mismo techo. Esto recalca la dificultad de emplear la representación directa para el cálculo grafico de los valores de Vmax y Km. Sin embargo, con la ayuda de un programa de regresión no lineal, es posible determinar un estimad de Km y Vmax a partir de esta representación, cuyo valor constituye un mejor estimado que el obtenido por cualquiera de los métodos gráficos descritos en la literatura, salvo quizá una excepción (ref).
b) los valores de Vmax y Km se obtiene de la serie realizada en ausencia de inhibidor (O); aquí, Vmax = 1/0.38 (mmol min-1 mg prot-1) = 2.63 (mmol min-1 mg prot-1) y -1/Km = -0.51 mM-1 ó Km = -1/-0.51 mM = 1.95 mM de glutamato.
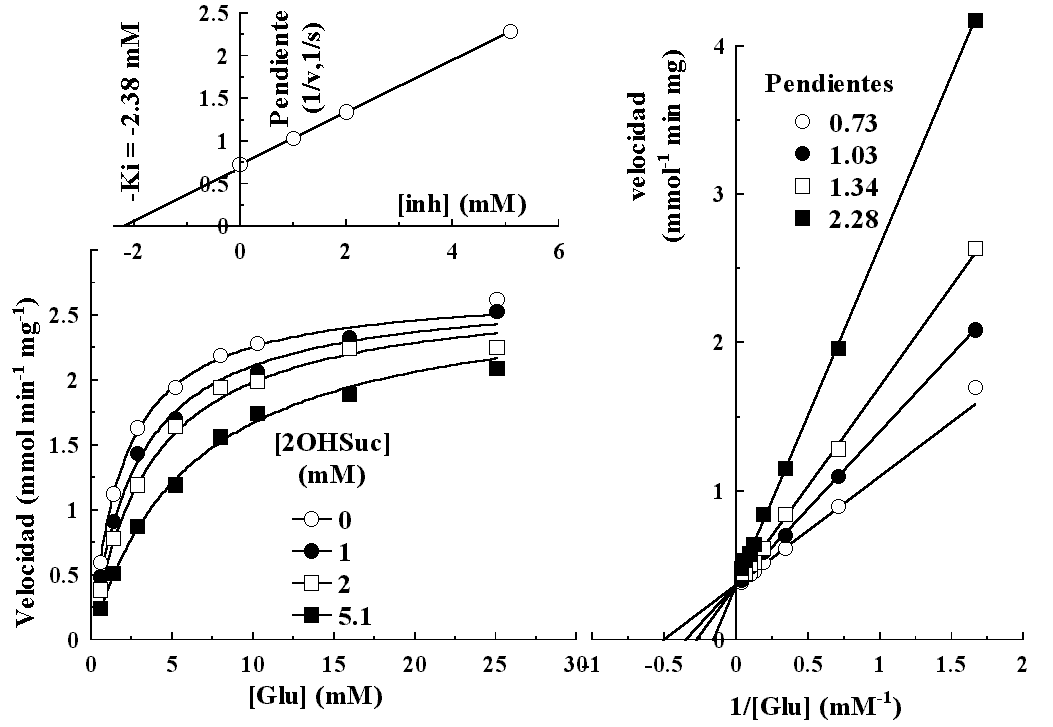
c) Finalmente, el valor de Ki puede obtenerse ahora del efecto que el inhibidor produce sobre la pendiente de la gráfica de Linewaver-Burk (que en ausencia de inhibidor es numéricamente igual a Km/Vmax). De nuevo, si se grafica el valor de estas pendientes frente a la concentración de inhibidor empleada (panel superior izquierdo). El valor obtenido para Ki es ahora 2.38 mM de 2-hidroxisuccinato. Es importante recalcar que cuando las rectas no se cortan en el eje "Y" como en este caso, esto significará que el inhibidor ( no competitivo) producirá alteraciones en los valores de la pendiente y de los interceptos con la ordenada del gráfico (1/v, 1/s), por tanto aquí se podrán obtener dos valores de Ki (usualmente llamados Kiu y Kic, respectivamente). Lo que representa una medida inversa de la fuerza con la que el inhibidor se une a la enzima cuando esta tiene su sitio activo vacío (Kic) o de la fuerza de unión a la enzima con el sitio activo ocupado (Kiu), por ello es que se obtienen dos valores de Ki. Si todas las rectas se cortasen en un punto sobre el eje "X", esto significaría que Kiu y Kic son numéricmente idénticos, pero esta coincidencia no es el caso más común.
----------regresar al índice de la sección -------------------regresar al índice general------------
11.- En este problema se presenta un caso común en la investigación farmacológica. Se trata de buscar un inhibidor que sea efectivo contra una enzima que resulta vital para un microorganimo y, por tanto, nos puede servir para defendernos de él. Lo primero que observamos, es que en lugar de hacer medidas a diferentes concentraciones de sustrato en presencia y ausencia del activador, aquí se realizaron determinaciones de la actividad a una u otra concentración fija de sustrato y con cantidades crecientes del inhibidor.
a) Para identificar el tipo de inhibidor que actúa, se pueden emplear diversas estrategias, por ejemplo, se pueden ordenar los datos de manera que se tienen dos valores de velocidad y dos concentraciones de sustrato para diferentes concentraciones de inhibidor. Esto es lo que se hizo en la figura inferior (panel B). Nótese que si trazaraos rectas estas cruzarían hacia el lado negativo del eje. Dado que las medidas de actividad se realizan sin añadir producto, una velocidad negativa carece de sentido, porque tendría que generarse sustrato en lugar de desaparecer y si no hay producto - ¿de dónde puede aparecer sustrato?
El resultado anterior nos indica que la cinética de esta enzima no es michaelina (hiperbólica), por lo que no podemos trazar rectas, sino que debería mos trazar curvas en esta figura. Por lo que lo único que podemos decir del tipo de inhibición con estos datos es que podría ser complejo y que se necesita más información para determinarlo.

b) Respecto al valor de I50, las curvas y los valores correspondiente se muestran en la figura inmediata superior, panel A. Como puede observarse el valor de I50 decrese cuando hay menor cantidad de sustrato. El valor de I50 nos indica cuanto inhibidor requerimos para inhibir la actividad de la enzima a la mitad en una cierta condición. Es evidente que el resultado nos dice que si hay más sustrato disponible el inhibidor será menos efectivo y refleja la existencia de un componente competitivo en la inhibición, lo que no significa que la inhibición sea realmente competitiva.
c) Como puede verse, si otros inhibidores se quisieran comparar con este, primero habría que asumir que el mecanismo de inhibición es el mismo. Esto suele hacerse en la investigación farmacológica, ya que a menudo los compuestos a ensayar son iguales en su estructura química, excepto por uno o dos substituyentes. En segundo término, los valores de I50 tendrán que haber sido determinados con la enzima ensayada en condiciones idénticas, excepto por la adición del inhibidor en estudio. Por ello, la respuesta a la pregunta que se plantea en el problema es NO - no es posible compara los valores puesto que no fueron medidos bajo las mismas condiciones.
----------regresar al índice de la sección -------------------regresar al índice general------------
Solución a los problemas sobre los mecanismos de catálisis.
Esta sección contiene 7 problemas y se encuentra aún en contrucción.
----------regresar al índice general------------
1.- El mecanismo de catálisis de la Ribonucleasa indica que hay tres grupos químicos escenciales para la catálisis de la hidrólisis del enlace ester de fosfato del RNA. Dos de ellos (la H12 y la H119) pertenecen a la enzima, mientras que el tercero el el propio hidroxilo de la posición 2 de la ribosa del RNA (el sustrato), el que, activado por la H119 (base general) se convierte en un nucleófilo activo capáz de atacar el átomo de fosforo iniciando así el rearreglo de diester fosfórico que lleva a la hidrólisis del RNA. Este mismo mecanismo puede ser catalizado por otras bases, incuído el grupo hidroxilo, de manera que se generen nucleótiods 2',3' fosfato cíclicos, con hidrólisis del enlace 5'fosfato adyacente. Por ello, el RNA se hidroliza fácilmente en medio ligeramente alcalinos (pH >8). El DNA no cuenta con el OH de la posición 2', ya que la azúcar que lo compone el la desoxirribosa. Consecuentemente es mucho más estable a pH's alcalinos. De hecho, esta caractraística se emplea para purificar el DNA, ya que al incubarlo a pH de 8 es posible eliminar el RNA que pueda venir como contaminante.
----------regresar al índice de la sección -------------------regresar al índice general------------
2.- En este caso, la presencia de grupos más voluminosos en la molécula forza al grupo carboxilo a aproximarse más al -OH fenólico, lo que favorece la lactonización. De hecho, la geometría de la molécula en tre dimensiones revela que a fin de acomodar los grupos tan voluminosos el anillo benzenico debe permitir una leve pero apreciable desviación de la planaridad, por consiguiente, la eliminación de un átomo de oxígeno como resultado de la lactonización hace más estable la geometría de toda la molécula.
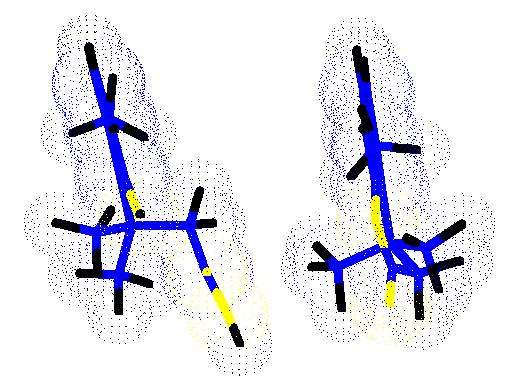
En la imagen anterior se puede ver como la posición de los metilos en la molécula (vista aquí desde el canto superior del anillo bencénico) favorecen conformaciones en las que hay mayor aproximación de los grupos carboxilato y OH fenólico (identificados aquí por el color amarillo de los átomos de oxígeno.
En las enzimas, la cudadosa orientación geométrica de los grupos químicos en el sitio activo de la enzima favorece la unión de los sustratos con geomietrías predeterminadas, de modo que se eleva notablemente la probabilidad de que los grupos que deben reaccionar se orienten correctamente. Estos se denominan factores de proximidad y orientación, siendo uno de los efectos importantes en la catálisis enzimática.
----------regresar al índice de la sección -------------------regresar al índice general------------
3.- La habilidad de una enzima para favorecer una reacción química y aumentar la velocidad con la que esta ocurre depende de que la barrera energetica que separa al estado de transición de los reactante pueda ser superada. Por ello, las enzima actúan empleando caminos químicos distintos a los que se observan en solución y modificando el entorno químicos de los reactantes y del estado de transión a fin de favorecer la ocurrencia de este último. Esto significa que la diferencia energítica entre reactantes y estado de transición debe reducirse. Pero si el complejo enzima sustrato al formarse libera una gran cantidad de energía libre, esto significa que el estado basal de los reactantes se encuentra a un nivel energítico aún más bajo, por consiguiente, alcanzar el estado de transición requeriría ahora mucha más energía, a menos que el complejo enzima-estado de transición sea todavía más estable con respecto al estado de transicón en solución, que lo que es el complejo enzima sustrato con respecto al reactante. Por ello, una enzima que realmente catalice una reacción debe estabilizar más el estado de transición que lo que estabiliza al sustrato al interactuar con cada uno de ellos. De otra manera, no habra reducción de la energía de activación efectiva y no se favorecerá la catálisis.
De hecho, una de las maneras en las que puede hacerse a una enzima peor catalizador, es modificando ciertos grupos importantes en la interacción enzima-sustrato de modo que la unión sea más fuerte, el resultado es una enzima que libera lentamente los productos y, en consecuencia, cataliza más lentamente la reacción, aún cuando si kcat (medida cinéticamente) cambie poco.

La figura anterior ilustra tres casos, la reacción no catalizada, la reacción en presencia de la supuesta enzima descrita en el problema y el caso de una verdadera enzima en la que el estado de transición es estabilizado mucho más que el sustrato al unirse con la enzima. En los dos primeros casos la energía de activación es la misma y por tanto no habra aumento en la velocidad de la reacción.
----------regresar al índice de la sección -------------------regresar al índice general------------
4.- Los estudios modernos sobre la catálisis enzimática demuestran que cambios en aminoácidos cuyo papel se considera de unión pueden resultar en alteraci´nes importantes en la kcat y viceversa. El papel de estos grupos es asignado casi siempre con base a la posición del grupo en los cristales de la enzima obtenidos en presencia de analogos del sustrato, datos que se compaginan con eltras evidencia experimentales diversas. Debido a que la geometría completa del sitio activo ha sido diseñada para favorecer una mayor estabilidad del estado de transición, aún pequeños cambios en la polaridad de los grupos y o su orientación pueden tener efectos importantes sobre la catálisis. Por ello es muy difícil asignar papeles específicos a los residuos de aminoácido delimitando su finción a únicamente unión y únicamente catálisis, de hecho, basta recordar que durante la catálisi los intermediarios deben permanecer unidos a la proteína, de otro modo no podríamos adscribirle la catálisi a la enzima.
Sin embargo, cabe aclarar que las determinaciones cinéticas demuestran que los eventos de unión de los sustratos al sitio activo de la enzima y los evéntos químicos de catálisis ocurren en ciertan secuencia y no son simultáneos. Es por tanto factible pensar que durante los eventos de unión hay grupos que no son importantes, sino hasta que se inicien los eventos catalíticos, en este sentido, quizá sea posible distnguir entre grupos que participan en unión y en catálisis y grupos que participan principalmente en catálisis.
----------regresar al índice de la sección -------------------regresar al índice general------------
5.- La tripsina y la quimotripsina son proteasas muy activas pertenecientes a la familia de las serín-proteasas. Estas proteínas difieren en la especificidad por el enlace peptídico que hidrolizan; mientra la tripsina es muy selectiva hacia enlaces peptídicos en los que participan aminoácidos básicos con carga positiva (Lys y Arg), la quimotripsina prefiere aminoácidos aromáticos y puede también atacar enlaces peptídicos en los que participen aminoácidos alifáticos voluminosos, principalmente L, I y V. En consecuencia, la quimotrispsina es menos activa frente a proteína nativas, ya que es menos probable que un enlace peptídico formado por aminoácidos apolares se encuentre en la superficie de la proteína, generalmente estos se encuentran en el interior. Por el contrario, la tripsina ataca aminoácidos polares y cargados, que con mucha frecuencia se encuentran en la superficien de la proteína, expuestos al solvente. Así, la quimotripsina es poco activa contra si misma en su estado nativo, mientra la tripsina es muy activa contra su propio estado nativo, por ello es una proteína difícil de purificar. Afortunadamente, son también proteínas que recuperan fácilmente su plegamiento nativo y es posible purificarlas a pH's extremos en los que su actividad es mínima y por tanto no se autodigieren, una vez purificadas se pueden incubar al pH en el que son activas para que recuperen su conformación nativa y sirvan a los diversos fines científicos y/o tecnológicos que requirieron su purificación.
----------regresar al índice de la sección -------------------regresar al índice general------------
6.- a) El inhibidor de tripsina de soya es una pequeña proteína capáz de unirse al sitio activo de la tripsina con gran fuerza, de modo que aunque es hidrolizado uno de sus enlaces peptídicos, la enzima no puede liberar el producto y esto evita que continúe con su actividad hidrolítica. Dado que la tripsina es una de las enzimas importantes en la digestión de las proteínas que nos sirven como alimento, es necesario inactivarlo a fin de que los productos elaborados con proteína de frijol soya puedan ser digeridos por los humanos. De hecho la ingestión de proteína de soya cruda puede causar la muerte por congestión digestiva.
b) En el caso del Gorgojo de la soya, es evidente que debe poseer proteasas especiales cuya especificidas y bolsillo de unión sean diferentes de la de la tripsina, lo que las hace insensibles al inhibidor.
----------regresar al índice de la sección -------------------regresar al índice general------------
7.- La respuesta a esta pregunta está en el cuidadoso balance entre rigidez y flexibilidad de la estructura de la proteína, que permite que cada grupo químico se encuentre casi contínuamente rodeado de un conjunto de aminoácidos definidos. Esto es epecielmente cierto en el sitio activo, en el que la disposición y ordenamiento especial de los grupos químicos está cuidadosamente elegida para favorecer la catálisis. De este modo, un protón, atrapado por un grupo químico con baja afinidad protónica puede encontrarse demasiado lejos de la base como para ser transferido a esta y con frecuencia, puede además estar estabilizado por la presencia de otros grupos con densidad electrónica, lo que favorece que permanezca en dicho entorno, en lugar de migrar hacia el entorno de la base, el que a su vez puede estar rodeado de grupos can baja densisdad electrónica reduciendo la estabilidad de los protones que entren en este sitio.
----------regresar al índice de la sección -------------------regresar al índice general------------